
Copyright (C) 2022, Hongfei Liu 2022/08/16
![]()
![]()
The goal of handyFunctions is to get rid of the barrier to deal with non-standard data.frame format for R newbies, especially the user in bioinformatics data analysis. Besides, there are also some required plot functions for downstream analysis of dataset generated from vcftools and plink.
You can install the development version of handyFunctions like so:
## Clone it from github and install it locally
git clone https://github.com/LuffyLouis/handyFunctions.git
## OR
## Install it in R
remotes::install_github("LuffyLouis/handyFunctions")handyFunctionspackage contain three main sections,
including ReformatDataframe, InteractDataframe, and Post-VCF. There are
some basic examples which show you how to solve common problems in data
analysis:
This section is designed to reformat data.frame with odd colnames, rownames, or even inappropriate dtypes for each columns.
Based on the following example unifyDataframe function,
you can change the formats of raw data.frame to what you want.
Especially for the dtypes in data.frame, you can set
custom=FALSE for automatically changing into appropriate
dtypes.
library('handyFunctions')
data("people")
head(people)
#> ..name ..sex ..age ..death..age
#> 1 Ming Li male 12 34
#> 2 Zixuan Liu female 23 thirty
#> 3 Yizhen Zhu male NA 54
#> 4 Lingling Wang female 21 77
#> 5 Bang Wei male 11 <NA>
#> 6 Xiaoyu Chen female 74 89
modifiedPeople <- unifyDataframe(people,rawColSep = '[.][.]')
head(modifiedPeople)
#> name sex age death_age
#> 1 Ming Li male 12 34
#> 2 Zixuan Liu female 23 thirty
#> 3 Yizhen Zhu male <NA> 54
#> 4 Lingling Wang female 21 77
#> 5 Bang Wei male 11 <NA>
#> 6 Xiaoyu Chen female 74 89Note: due to the separation supporting regEx, please
use the "[.][.]" for reformatting people
data.frame.
The InteractDataframe section is designed for
interaction between two data.frame or one data.frame and another
vector.
Sometimes, we often find it fuzzy and tedious while we’d like to
merge two data.frame with different colnames. Therefore,
mergeCustom function may be the better solution to get it
rid of.
library('handyFunctions')
data("people");data("grade")
head(people)
#> ..name ..sex ..age ..death..age
#> 1 Ming Li male 12 34
#> 2 Zixuan Liu female 23 thirty
#> 3 Yizhen Zhu male NA 54
#> 4 Lingling Wang female 21 77
#> 5 Bang Wei male 11 <NA>
#> 6 Xiaoyu Chen female 74 89
head(grade)
#> name chinese math english physics biology chemistry
#> 1 Ming Li 120 130 145 80 90 99
#> 2 Zixuan Liu 109 120 110 85 99 88
#> 3 Yizhen Zhu 98 113 100 74 100 76
#> 4 Lingling Wang 138 145 126 55 89 100
#> 5 Bang Wei 119 105 139 100 78 99
#> 6 Xiaoyu Chen 119 105 120 69 80 77
merged <- mergeCustom(people,grade,xcol = '..name',ycol = 'name')
head(merged)
#> ..name ..sex ..age ..death..age chinese math english physics biology
#> 1 Bang Wei male 11 <NA> 119 105 139 100 78
#> 2 Lingling Wang female 21 77 138 145 126 55 89
#> 3 Ming Li male 12 34 120 130 145 80 90
#> 4 Xiaoyu Chen female 74 89 119 105 120 69 80
#> 5 Yizhen Zhu male NA 54 98 113 100 74 100
#> 6 Zixuan Liu female 23 thirty 109 120 110 85 99
#> chemistry
#> 1 99
#> 2 100
#> 3 99
#> 4 77
#> 5 76
#> 6 88library('handyFunctions')
library('ggplot2')
data("SNV_1MB_density_data")
head(SNV_1MB_density_data)
#> CHROM BIN_START SNP_COUNT VARIANTS.KB
#> 1 1 0 253 0.253
#> 2 1 1000000 31 0.031
#> 3 1 2000000 208 0.208
#> 4 1 3000000 77 0.077
#> 5 1 4000000 204 0.204
#> 6 1 5000000 75 0.075
ShowSNPDensityPlot(SNV_1MB_density_data,binSize=1e6,chromSet = c(38:1),withchr=T)+
theme(axis.text.y = element_text(size=12))
#> ## Filtering the density data with specific chrom set...
#> ## Judging if it should be added the chr and factoring...
#> ## Reformatting the raw density data...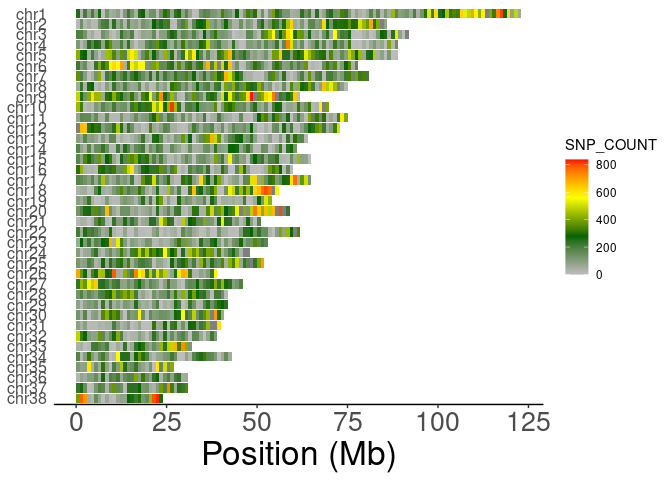
This project is published under MIT license, see file
LICENSE.